


Hypertension artérielle… Et si c’était un phéochromocytome ?

Souffrir d’une hypertension n’a rien d’exceptionnel. Souvent le fruit d’une hygiène de vie défaillante, sans parler de l’hérédité et de l’âge, l’hypertension artérielle peut également être liée à une pathologie sans rapport avec le style de vie. C’est le phéochromocytome, une tumeur qui sécrète des hormones hypertensives.
Selon l’OMS, un adulte sur trois souffrirait d’une hypertension artérielle (HTA) dans le monde. En France, on dénombre environ 14 millions d’hypertendus dont beaucoup s’ignorent. Près de la moitié des décès par accident vasculaire cérébral et cardiopathie serait liée à cette maladie cardio-vasculaire. D’où l’intérêt d’un dépistage et d’une prise en charge active. Mais si la survenue de l’HTA est largement conditionnée par le style de vie, il est une pathologie sournoise qui explique à elle seule près de 1 % des HTA : c’est le phéochromocytome*, autrement dit une tumeur, bénigne ou maligne (10 % des cas), qui concerne les glandes surrénales dans 90 % des cas. Le phéochromocytome ne concerne que les adultes entre 20 et 50 ans, sans distinction de sexe.
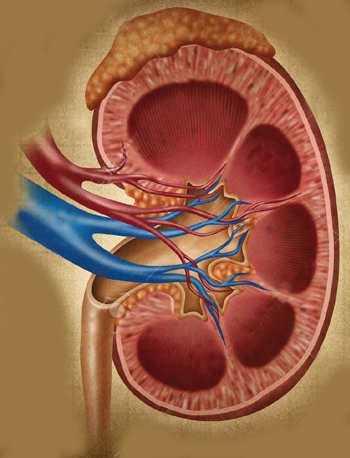
Glandes surrénales
Les glandes surrénales ne payent pas de mine, du fait de leur petite taille (3 cm de long pour 2 cm de haut et 1 cm de large) et de leur poids plume (4 à 6 g en moyenne). Et pourtant. Situées au-dessus du pôle supérieur des reins, les glandes surrénales sont essentielles à la vie car elles synthétisent de très nombreuses hormones. Plus précisément, les surrénales sont constituées de deux parties, la corticosurrénale en périphérie de la glande (sécrétion de cortisol, d’aldostérone, d’hormones sexuelles) et la médullosurrénale au centre (sécrétion d’adrénaline et de noradrénaline). C’est justement dans la médullosurrénale que se développe le phéochromocytome. Cette tumeur se met à synthétiser de grandes quantités d’adrénaline et de noradrénaline, qu’on appelle aussi les cathécholamines, à l’origine de l’augmentation de la tension artérielle et de l’accélération du rythme cardiaque. Dans 10 % des cas, le phéochromocytome se développe en dehors des glandes surrénales, par exemple dans l’abdomen près de l’aorte, autour de la vessie ou dans le thorax.
Une poussée tensionnelle à haut risque
Plus qu’une simple HTA chronique, le phéochromocytome se caractérise par une poussée hypertensive aiguë sévère, au repos ou déclenchée par l’effort, de simples mouvements, les émotions, certains aliments ou des médicaments. Elle est accompagnée de sueurs, de palpitations et surtout de céphalées. La crise est souvent suivie d’une envie pressante d’uriner. Ce type d’HTA constitue une urgence. En effet, cette poussée tensionnelle aiguë qui peut durer une heure va menacer le cœur (risque d’infarctus), le cerveau (risque d’accident vasculaire cérébral ou d’hémorragie), les poumons (risque d’œdème du poumon), à court terme comme de façon chronique, ainsi que les yeux (développement d’une rétinopathie). Car dans certains cas, l’hypertension s’aggrave encore lors des poussées.
Dosages sanguins et urinaires
Difficile de diagnostiquer avec certitude un phéochromocytome sur la seule constatation d’une crise hypertensive.
Pour autant et lorsqu’on y pense, il est possible de repérer l’existence de cette tumeur par un dosage sanguin de l’adrénaline.
On peut aussi retrouver de grandes quantités d’adrénaline, ou ses dérivés (métanéphrines urinaires), dans les urines collectées sur 24 heures. En cas de phéochromocytome, les métanéphrines urinaires sont élevées.
Reste ensuite à déterminer où se situe la tumeur en explorant tout d’abord les glandes surrénales (90 % des atteintes), à l’aide du scanner ou de l’IRM.
Lorsque le scanner et l’IRM ne retrouvent pas de lésion surrénalienne, il s’agit d’un phéochromocytome dit « extra-surrénalien » qui nécessite alors le recours à la scintigraphie pour localiser le siège exact de la tumeur (bassin, abdomen, thorax…).
Chirurgie
Cette HTA menaçante nécessite un traitement médicamenteux intensif de toute urgence. Malheureusement, la crise hypertensive liée à un phéochromocytome résiste souvent aux traitements habituels. Le seul traitement réellement efficace, immédiat et radical, consiste en l’ablation chirurgicale de la surrénale concernée. Un acte chirurgical risqué, et donc effectué avec prudence, car la tumeur peut émettre une grande quantité de catécholamines lors de son retrait, exposant le patient à une crise hypertensive soudaine et malvenue. Une surveillance est indispensable dans les années qui suivent l’ablation si le phéochromocytome retiré est cancéreux. Ce n’est que lorsque le phéochromocytome est bilatéral (ablation des deux glandes surrénales), qu’un traitement substitutif devient nécessaire afin de compenser la perte des hormones synthétisées par les deux glandes.
Prédisposition génétique
Dans près de 10 % des cas, le phéochromocytome est d’origine génétique et peut s’associer à d’autres pathologies telles que :
- La maladie de Recklinghausen (neurofibromatose), qui se manifeste par des taches cutanées de couleur « café au lait » et des atteintes neurologiques.
- La maladie de Von Hippel-Lindau, caractérisée par des tumeurs vasculaires au niveau rénal et rétinien.
- Les néoplasies endocriniennes multiples (NEM), où le phéochromocytome peut s’associer à une hyperparathyroïdie (exagération du fonctionnement des glandes parathyroïdes, situées près de la thyroïde) ou à un cancer de la thyroïde.
Autres causes possibles d’hypertension
- Abus de sel alimentaire
- Surpoids
- Stress
- Grossesse
- Anxiété
- Tabagisme
- Sédentarité
- Hérédité
- Âge
- Insuffisance rénale
- Apnées du sommeil
- Sténose de l’artère rénale
- Hyperplasie des surrénales
- Hypercortisolémie
- Hyperthyroïdie
* Le phéochromocytome a été découvert en 1886.


